tRex is a comprehensive, data-driven online resource specifically dedicated to tRNA-derived fragments in the model plant Arabidopsis thaliana from various tissues, ecotypes, genotypes and stress conditions. The portal is based on verified Arabidopsis tRNA annotation and includes in-house generated and publicly available sRNA-seq experiments.
Users can explore data from 105 various genotypes, 5 ecotypes, 13 developmental stages and 7 tissues of Arabidopsis thaliana for a total of 300 samples and 42 experimental sets.
To retain compatibility with other resources dedicated to tRFs (e.g., tRFdb, MINTbase and tRF2Cancer) and to address subtle differences between these sequences we have enhanced the tRF classification. In tRex the tRFs can belong to any of ten distinct classes.
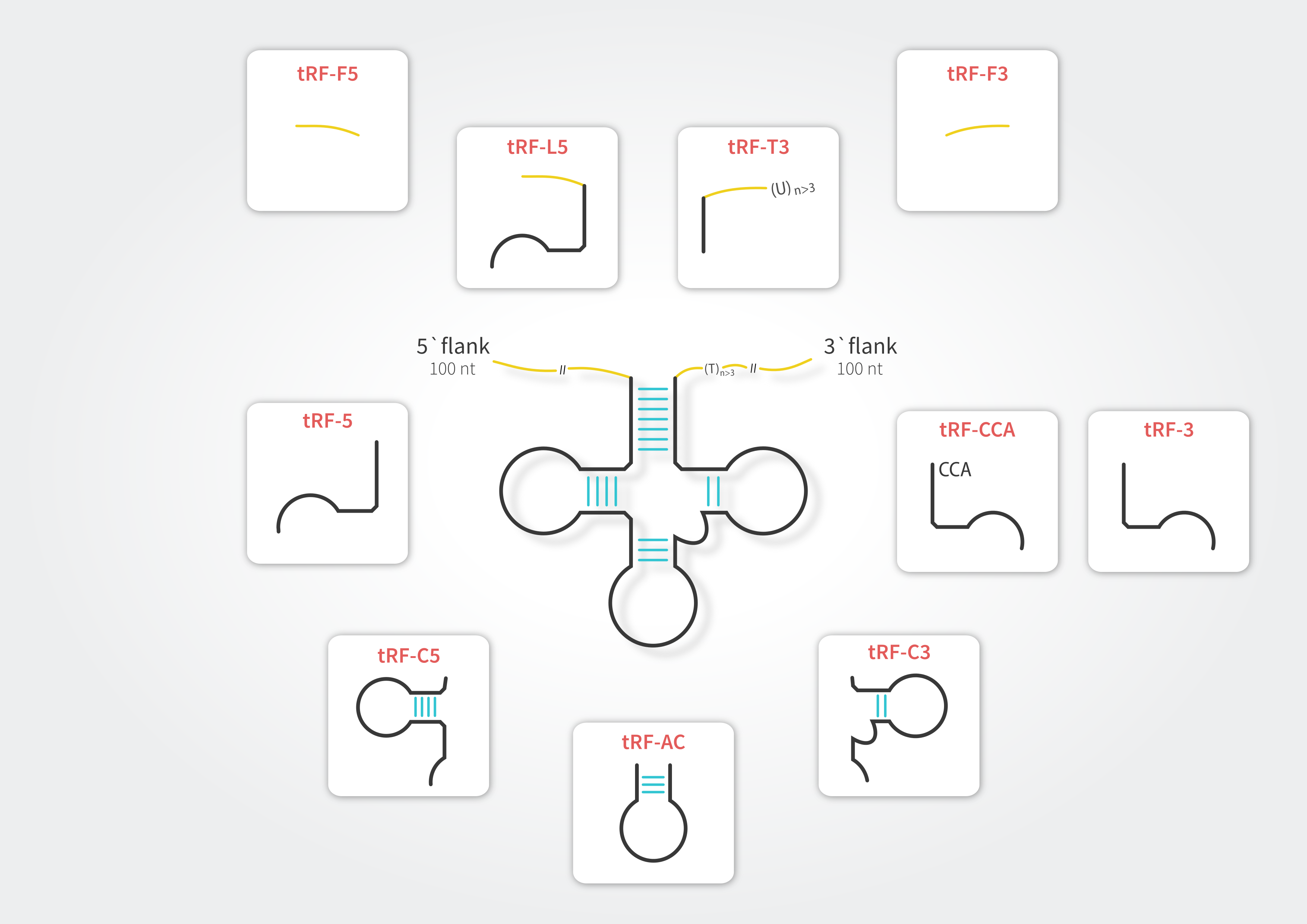
tRNA-derived fragments in tRex database refer to all tRNA-mapping reads with lengths between 15 and 28 nucleotides. Considering the fact that tRNAs undergo extensive post-transcriptional modification processes during maturation, we allowed for presence of multiple mismatches in the sequence alignment. To filter out potential false positive signals, we mapped to both tRNA reference set and Arabidopsis thaliana genome and discarded those sequences which mapped better (showing higher similarity) to loci other than tRNA. Each fragment is classified into one of the tRF types in accordance with its alignment position on the source tRNA. Due to considerable sequence redundancy among tRNA genes, a single fragment may enter multiple tRF categories.
The tRex database can be accessed via any of the three basic procedures:
- Selecting a tRNA
- Selecting a Sample
- Employing a similarity sequence search
The first two provide options to select one or multiple entries in any combination, using especially designed tables that allow for convenient filtering of data (utilizing checkmarks). Additionally, the tables can be sorted and searched. Multiple selection is implemented by using a shop-like method, where the user can add selected tRNAs or Samples into a “cart” . The content of the basket can be modified by selecting icon. Depending on the chosen option, the data can be presented as a single page record (tRF/tRNA/Sample) or expression profile (Full/Simplified/Heatmap).
To access information regarding a particular tRF, the user can search the database content using a sequence of interest. The database is equipped with 3 BLAST algorithms (megablast, blastn and blastn for short sequences). The user can access details about identified tRF or tRNA sequences directly from the BLAST results page.
At each stage of results exploration, the user has an option to access detailed information about a particular sample, tRNA and expression profile (full profile, structural profiles and read distribution).
The coverage profiles presented in tRex are calculated for raw counts as well as three different normalization methods:
- tRNA-based - counts are obtained by multiplying the raw count by 0.5 million and dividing by the sum of all reads mapping to tRNAs within a sample;
- Sample-depth-based - raw counts are multiplied by 1 million and divided by the sum of counts of all genome mapping reads within a sample
- redundancy-based - sample-depth normalized read counts are divided by a number of unique tRNAs to which a read is mapping.
tRF identification procedure
The annotation procedure involved mapping of each read to the reference tRNA dataset and the Arabidopsis genomic sequence allowing up to 20% of mismatches. This information was subsequently used to select the best sRNA - tRNA pairs and filter (remove reads mapping outside of tRNA space) as well as normalize (recalculate reads counts) of sRNA-seq data. The procedure was performed with Bowtie2 and samtools programs along with in-house generated Perl and Python scripts.
In order to account for the CCA end as well as potential modified nucleotides, we have adjusted default mapping parameters and used the following command:
Bowtie2-align -f --end-to-end -a -N 1 -L 8 --score-min L,-0.9,-0.9 -p X -x [tRNA reference index file] -U [file with all small RNA reads] -S [output file].
After the mapping we calculate a score for each alignment based on the number of mismatches, insertions and deletions. In cases when a read maps to the genome with a better score than to the “tRNA space” it is removed from the pool of candidate tRFs and not included in the tRex portal.
RNA structural analyses
The sequences of annotated tRNA genes and genomic regions showing high sequence similarity to tRNAs identified by blast were initially aligned using clustalw. The alignments were edited by hand to produce structural alignments reflecting the standard cloverleaf structure of tRNA. The structural alignments were used to design a template for displaying the tRNA structure diagrams.
The minimal free energy (MFE) structures were calculated using RNAshapes and the MFE diagrams were produced using VARNA software)
Modification predictions
The modified nucleotides were predicted using HAMR software (Ryvkin et al. 2013) with recommended parameters: minimum sequencing quality = 30, minimum read coverage = 10, expected percentage of mismatches due to sequencing error = 0.05, hypothesis to be tested = H4, maximum p-value cutoff = 0.01, maximum FDR cutoff = 0.05 and minimum percentage of reads to match the reference nucleotide = 0.05.
Target predictions
Putative targets were predicted using in-house program hybrid-blast (Szymanski M. unpublished). The obtained results were analyzed using RStudio version 1.0.136 and filtered based on a minimum threshold of 70% of perfect hybridization energy predicted with RNAfold software from the Vienna package. tRex database reports up to 50 of top alignments.
If you make use of the data presented in tRex, please cite the following article:
[..]